





Depicting tumor clonal evolution
- Jun 5, 2018
- 3 min read
Tumor clonal evolution
Cancer is stubborn largely due to its nature of heterogeneity: they evolve to adjust the changes of surrounding environment.

Figure above shows tumor clonal evolution. During the long lasting tumorigenesis period, tumor generates heterogeneous clones that can resist tumor suppressing factors such as the immune surveillance or apoptosis. During therapeutic period, some tumor clones escape from being targeted by, for example, not presenting the target. Therefore, obtaining a whole picture of how tumor clones evolve is crucial for establishing effective therapeutic strategy.
Depicting clonal changes with SciClone
With the advance of high throughout sequencing technique, a number of computational tools emerge to shed light on tumor clonal changes. Most existing tools, such as PyClone / ABSOLUTE, use Markov chain Monte Carlo ( MCMC ) to make clonality inference based on copy number data ( CNV ) and variant allele frequency (VAF) from temporally or spatially distinct samples. SciClone, however, takes variants within copy number neutral region (diploid) and perform clustering based on VAF using variational Bayesian mixture model. "variantiional" here means that a prior distribution of dirichlet process is defined to allow the algorithm determine the n_cluster by itself. SciClone only use somatic variants within diploid region to eliminate the VAF bias due to ploidy variation. Therefore the overall accuracy of SciClone depends on the accurate CNV estimation as well as the amount of high quality somatic variant.
Clonal evolution of leukemia before / after transplant
Here we make demonstration of clonal evolution of leukemia sample before and after transplant. We used MuTect2 and VarScan on Leukemia WES data to obtain somatic variants and CNV. During clonal inference, A total number of 320 out of 429 somatic variants were accepted by SciClone after excluding variants within copy number alteration region and variants with low depth of coverage. The SciClone paper recommends minimum input variant of 200 for accurate inference.

Figure above shows how the result looks like. For pre-sample (before transplant) on the left side, the top figure shows the VAF density distribution. x-axis represents the VAF and y-axis represents the amount of variants with corresponding VAF. We can see that most of the variants are roughly clustered in two peaks (green curve). One peak at around VAF 50% represents primary clone (theoretically, heterozygous somatic variants within copy number neutral region of a relatively pure tumor have VAF of slightly less than 50%). The model fit / component fit ( grey curve ) shows three subclones clustered within VAF 0% - 30%. The left bottom figure consistently shows one primary clone and three subclone with corresponding VAF. Figure in right is the clonal result for post-sample (after transplant). it got a very much similar clonal pattern as pre-sample except that one subclone (orange) moved to the right. This comparison implies that the treatment didn't eliminate any tumor clones. In addition, one subclone even expanded. The 2D figure below offers a better view in which all clones lay diagonally except clone 2.
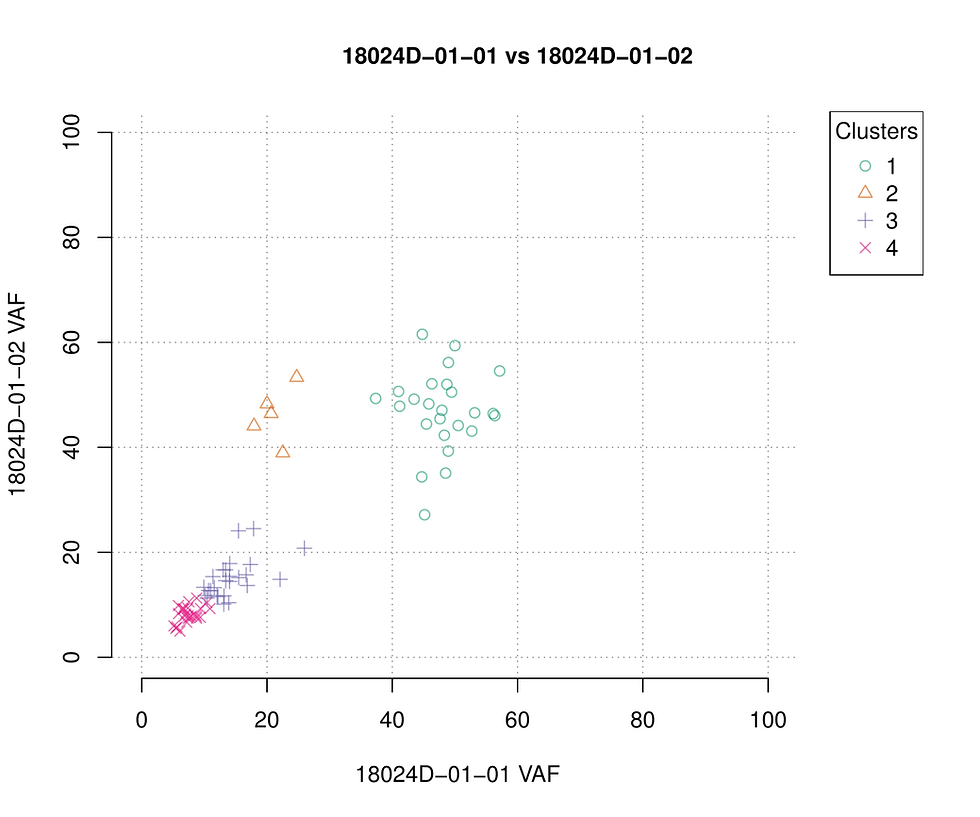
We then used WGS data of same samples to try to replicate the results. The amount of input somatic variants increases to over 30000. Apparently, SciClone failed to converge the cluster with such large amount of variants. Since the author claims the minimum input variants of 200 and we successfully performed the clustering with over 300 variants from leukemia: a relatively low mutation burden cancer type, we conclude that WES data is more suitable for SciClone.
As targeted therapy / immunotherapy advances, such clonal changes profile could be a key component for precision medicine. Now many biotech companies / research labs spend efforts on establishing ctDNA based liquid biopsy method for monitoring the blood concentration of specific variant. This "bulk analysis", although being invasive, can be an alternative to interrogate the clonal / VAF changes of large number of variants at once.
Comments